

背景及概述[1]
2-溴甲基-4-氟甲酸甲酯中文别名N,N,N',N'-四甲基-1,3-丙二胺、1,3-双(二甲氨基)丙烷,密度1.534g/cm3,沸点291.6ºC at 760 mmHg),闪点130.1ºC,折射率1.541,蒸气压0.00193mmHg at 25°C。2-溴甲基-4-氟甲酸甲酯可用作医药化工合成中间体。
制备 [1]
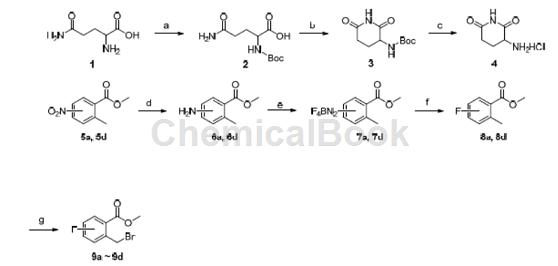
步骤a:N-(叔丁氧羰基)-L-谷氨酰胺(2)的合成
14.6 g L-谷氨酰胺溶解于100.0 mL 1.0 mol/L的NaOH 溶液,冰浴下缓慢滴加21.8 g 二碳酸二叔丁酯。撤走冰浴,室温下搅拌过夜。反应结束后用氢钾调pH=1~1.5,用500 mL 分3 次萃取,合并有机相,用无水钠干燥,减压旋干,得19.5 g 油状物,产率79%。可直接投下一步。
步骤b:N-(2,6-哌啶二酮-3-基)氨甲酸叔丁酯(3)的合成
19.0 g 化合物2、17.8 g EDCI 和12.6 g HOBt 溶于200 mL 乙腈中,室温下缓慢滴加15.7 g 三乙胺。将反应溶液转移至油浴中回流24 h。反应结束后,旋干溶剂,加入500 mL 水,室温搅拌1 h,抽滤,用大量清水洗涤滤饼,再用少量无水乙醇冲洗,抽干,得7.2 g 白色固体,产率41%。m.p. 214.4~214.5 ℃;
步骤c:3.2.3 3-氨基-2,6-哌啶二酮盐酸盐(4)的合成
用5.0 mL 1,4-二氧六环将1.4 g N-(2,6-哌啶二酮-3-基)氨甲酸叔丁酯溶解,加入30 mL 2.0 mol/L的1,4-二氧六环盐酸溶液,室温下搅拌4 h. 过滤,用少量洗涤滤饼,抽干,得0.99 g 白色固体,产率98%。m.p. 278.6~279.3 ℃;
步骤d:5d的合成将
9.0 g 2-甲基-3-硝基甲酸溶于40 mL 无水甲醇中,缓慢滴加15 mL SOCl2,室温下搅拌反应0.5 h,再转入油浴中回流1 h。反应结束后,旋干溶剂,加入200mL,分别用饱和Na2CO3 溶液和饱和食盐水溶液洗涤有机相1 次,分液、干燥;浓缩有机相,重结晶得9.1 g 浅黄色固体,产率93%。m.p. 65.5~66.1 ℃。可直接投下一步反应。
步骤e:2-氨基-6-甲基甲酸甲酯(6d)的合成
10.0 g 化合物5d、3.2 g 氯化铵和9.0 g 锌粉置于200 mL 乙醇/水(V/V=1/1)溶液中,65 ℃油浴反应。TLC监测反应直到原料反应完全。旋蒸出大部分溶剂,用400 mL 萃取,干燥有机相,浓缩,用浓盐酸成盐。抽滤,并用洗涤滤饼至白色,真空中干燥,得8.8 g 白色固体,产率85 %。
步骤f:2-氟-6-甲基甲酸甲酯(8d)的合成
8.5 g 化合物6d 悬浮于32.4 g 40%的四氟硼酸之中,冰盐浴下搅拌10 min. 用尽量少的水将3.5 g 亚硝酸钠溶解,缓慢地滴加入上述混合溶液中,继续搅拌1 h. 抽滤,用DCM 洗涤滤饼至浅黄色. 抽干,得7.9 g 固体。m.p. 89.2~90.0 ℃.
将7.9 g 上述所得固体悬浮于100 mL 中,缓慢升温至80 ℃,恒温后再小心升温至85 ℃,这时有大量白烟产生. 当白烟几乎消失后,再次升温并回流20 min.反应结束后,冷却至室温,加入100 mL Na2CO3 溶液,分液; 干燥有机相,浓缩,柱层析分离[洗脱剂: V(石油醚)∶V()=50∶1]得2.5 g无色油状物,产率24%。
步骤g:2-溴甲基-6-氟甲酸甲酯(9d)的合成
550 mg 化合物8a、695 mg NBS 和40 mg BPO 溶于30 mL CCl4中,油浴中回流6 h. 反应结束后,旋干溶剂,加入100 mL ,分别用饱和NaHCO3 溶液和饱和食盐水溶液洗涤有机相1 次,分液。干燥有机相,浓缩,柱层析分离[(洗脱剂:V(石油醚)∶V()=50∶1]得化合物2-溴甲基-4-氟甲酸甲酯,无色油状物,产率23%。
主要参考资料
[1] 陈衍炽, 任玉杰. α-(异吲哚啉酮-2-基) 戊二酰亚胺含氟类似物的合成与白血病细胞 K562 抑制活性[J]. 有机化学, 2015, 35(5): 1123-1130.


